
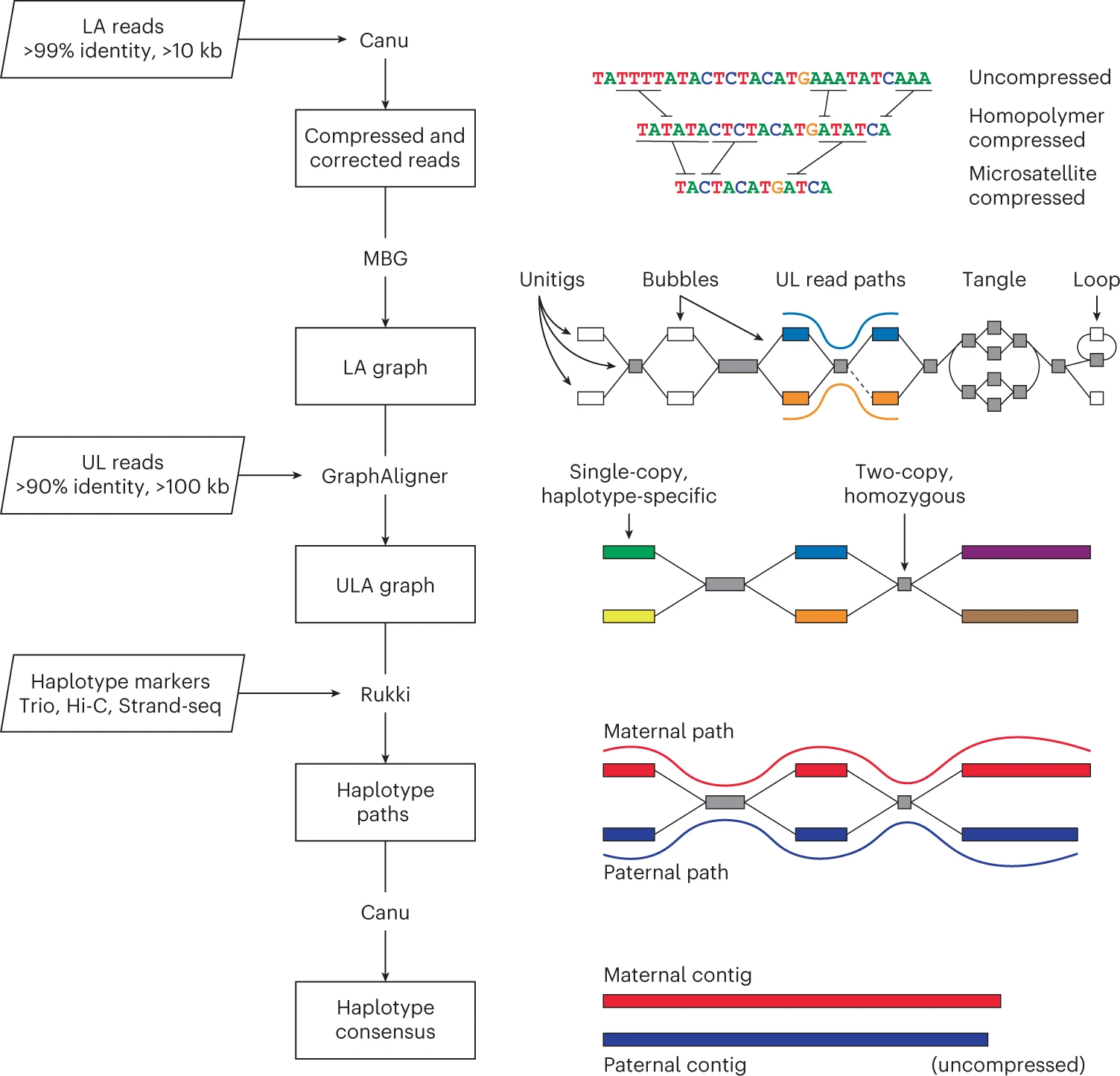
LA reads: long accurate reads
UL reads: ultra-long reads
问题, 重复序列压缩后,如何恢复成原来的序列
Methods
Error correction and homopolymer compression
根据hicanu及额外的提升准确率的方法对LA reads 纠错
将LA reads 中的同聚物压缩成单个碱基 AAAAAA-> A
将read all-vs-all 比对,如果一个read有一个位置被很多read 覆盖,但大部分的其他read在这个位置支持不同的碱基,read的这个位置被认为是错误,然后会被修正,如果至少两个read支持这个read上的碱基,就会保留这个碱基。
在后续所有步骤中都是用,修正后的同聚物压缩后的LA reads,只会在最终生成consensus的步骤恢复原来的形式。
Microsatellite compression
unit 单个长度2-6bp的序列, 例如 ACGACGACG 是3个ACG unit 重复3次, 用MBG进行微卫星压缩掩盖这些错误。
MBG使用的微卫星压缩类似同聚物的压缩。每个repeat unit用一个unique的字符表示,所有的串联重复被合并到一起,MBG将输入read转化成特殊的字母序列,一个单元,最长6bp,重复2次以上会被检测成微卫星,通过 unit seqence,unit length,overhang length 三个属性编码。overhang length 是微卫星序列段点上,不完整的unit的长度(ACGACGAC ,AC是overhang)overhang length 可能为0。 给定长度为n的unit,有n4^n种可能, unit 长度最长为6 ,少于2^16,可以用16位 integer表示unit字母表
根据overhang length对反向互补序列旋转,微卫星可以表示为 (x)y ,x是unit,y是overhang,例如 ACGACGACGAC 为 (ACG)AC, 反向互补是(GTC)GT。
选择最短的重复单元编码,被包括在其他unit的重复会被丢掉,CGTGTCGTGT 保留 (CGTGT) 丢弃 (GT)。微卫星之间可能会有重叠,裁剪重复边界来避免重复。ACGACGACGTCGTCGT 表示为ACGACGA和TCGTCGT,将CG裁剪掉,认为是无重复序列。
构图后,通过read对重复序列计算consensus来拓展压缩后的字符串。将read 编码成ACTG和表示repeat unit的16位整数,和一个map,表示整数和unit序列的对应关系。从所有reads中收集支持信息, 选择出现频率最高的作为consensus,如果存在相同的选择字典序大的。(这里应该是将read比对到图上,根据比对信息获取consensus吧)
Multiplex de Bruijn graph
MBG是一个用LA reads构建图的工具
给定一个kmer长度k,所有长度k的节点都是潜在可解析的,通过穿过节点的路径,对潜在可解析的节点查找跨越三元组triplet,一个节点n的跨越三元组是一个长度位3的子路径,中间节点是n,对于每个潜在可解析的节点,查找支持每个跨越三元组reads的数量。给定threshold t,如果支持的read超过t,称为solid三元组,没有solid triplet支持的边会被移除,一个所有边都没有solid triplet支持的顶点被标记为不可解析。当k小于4000时,禁用这个检查(不是大于么,应该是长度越长,出现错误的概率越高吧)。如果一个solid triplet 第一个或第三个节点被标记为不可解析,移除这个solid triplet,重复执行,将节点标记为不可解析和移除solid triplet这两个步骤,直到收敛(不变),将潜在可解析节点的集合标记为可解析的。
删除所有可解析的节点,创建k+1mer的edge-nodes。如果一条边连接一个可解析节点,edge-node长度为k+1,序列包括可解析node和另一个node的一个base。如果连接两个可解析节点,edge-node序列为两个node的路径的序列,edgenode根据solid triplet连接, n1n2n3,在n1n2,n2n3的两个edge-node节点之间天机一条边。重新根据这些新节点创建read path ,合并无分支路径。
在k增加的时候,MBG会清理图,在每个解析步骤,会清理tip尖端和交叉连接,尖端是仅在一段有连接的节点,如果尖端较短(<=10kb)且覆盖率低(<=3),如果移除他们不会生成新尖端,移除。交差连接是指错误连接两个基因组区域的节点。如果节点两侧都只有一条低覆盖率边的节点被视为可能的交叉连接。如果移除这个节点不会产生新尖端,移除。之后合并非分支路径。
重复执行 增加k和清理图的步骤,直到没有可解析节点或者k=15000。开始使用覆盖深度threshold t=2,之后用t=1解决更低coverage的区域。图上有错误生成的gap会用read路径修复,生成最后的LA graph。
UL read to graph alignment
将ULread比对到LA graph 填充gap和解决重复。
如果UL read 有一段alignment的结束一个尖端,另一个alignment的开始也是尖端,这个UL reads 支持这两个尖端之间的gap填充。如果一对尖端至少有3个UL reads支持填充,插入一个gap node到graph中,连接这两个尖端。gap的length有所有支持的UL reads的gap length的中值确定。如果gap是正值,gapnode的序列为相应数量的N,如果是负值,序列由尖端node复制过来,边标记为有overlap
用所有长度>=100kb的node估计基因组的平均coverage,所有node的coverage和这个平均coverage比较,长的并且coverage接近的标记为unique。然后coverage接近,长度不限的,如果路径一致标记为unique。路径一致定义为,如果一个节点上,80%的UL read 路径是相同的,或者互为后缀或前缀(UL read 和 节点序列是一致的)。在图上发现bubble结构。bubble可以分成单拷贝,双拷贝或者多拷贝。基于核心节点(bubble开始分裂的节点)。单拷贝的核心节点标记为unique,对于双拷贝链,如果bubble有两个路径,有类似的coverage并且类似于一般的chain coverage,bubble nodes 标记为unique,忽略多拷贝。
标记unique节点后, 开始使用UL reads构建unique顶点之间的路径。bridge 连接两个unique顶点,且之间没有unique顶点。UL 比对的路径为bridge。如果两个路径之间只有一个公共 unique node,这两个bridge认为是不一致的(?不懂)。这种情况,只考虑bridge的端点,而不是整个路径,如果一个不一致的bridge只有不到一半的read支持另一个bridge,这个可能是错误的,会被移除。如果没有任意一个bridge是其他bridge coverage的两倍,都保留。在每对 unique node之间有最多read支持的路径被作为consensus bridge路径。所有的至少有一半consensus 路径coverage的其他路径都被保留下来,丢掉其他低coverage路径。
最后,如果与table接触的所有 unique node 都有bridge,tangle 中所有的高coverage node 和边 被bridge覆盖。这些tangles可通过基于bridge连接unique顶点解决。可解决的tangle中的没有被路径覆盖的顶点认为是错误的,并被移除,被多个路径覆盖的 nonunique 顶点在相应路径中是重复。不可解的tangle没有unique node,或者这些unique nodes被识别错了。
连接完unique顶点后,用MBG中的算法利用UL read的路径处理图。
Graph cleaning
通过UL解图完成后,移除尖端。如果从一个分叉开始至少有一个长路径,其他从这个分叉开始的段路径可能会被移除,默认,long path >=35 kb, 裁剪的路径要么<=35kb要么 <10%长路径的长度。
每一步解决过程中都会输出简化路径中的节点和原来图的关系,这个顶点映射可以用来发现那些顶点在原来的MBG图是被包含的。可以用这个信息生成后续图上每个node的coverage估计。一个节点的coverage估计定义为包含的MBG图中节点的coverage的带权均值,最终图中只考虑包括的MBG节点。
这些coverage估计信息,用来解决序列错误引起的bubble结构,如果一个bubble chain是单拷贝的,组成它的bubble一定是测序错误引起的,保留高coverage的路径,丢掉路径外的节点和边,最终的图由 LA 和UL read 构成成为ULA graph
Haplotype reconstruction
Rukki 是一个从标签后的组装图提取单倍型的工具,它主要的通过分析单倍型特定的marker进行二倍体基因组的单倍型组装。目前,主要基于三个样本的单倍型重建,用父母illumina read 中识别的亲本特异性kmer作为单倍型标记
Rukki 根据单倍型marker的相关性将图上的节点标记为父系或者母系,如果marker是模糊的就不标,考虑node上marker数量的绝对值,平均密度和两个单倍型marker 数量的比值。Rukki还检测来自两个单倍型丰度都很高的节点,这些可能是错误的组装或连接了错误的不同单倍型。
长度大于500kb的node称为long node。这些long node 很可能是单拷贝或者单倍型特异性的。Rukki将 不同标签区域标记节点汇聚的邻居 标记为纯和。由于错误,纯和的节点可能会含有少量的亲本特异的marker,可能会被错误的标记,所以,未标签的和之前标签的但有升高的coverage(与之前得到的所有long node 的加权coverage相比)的node,都会被检测。剩余的long node 勇于启发式的拓展单倍型路径, 父系的节点和源自母系节点的路径不兼容,反之亦然,纯和节点和未标记节点可以加入任何路径。
为了使拓展更稳定,将长节点合并到路径时,会分析这个长节点的邻居,尝试识别下一个长候选节点。考虑边界为long read的子图,s是source node,如果source node和sink node都是标记的,并且存在和当前路径的单倍型 兼容的唯一sources,对应的sink标记为下一个长候选t。如果找到了,Rukki会进一步约束启发算法,优先考虑s到t的任何路径的节点。只考虑前向方向,反向方可以通过反向互补的前向得到。单倍型路径拓展可以推断出一些错过的单倍型标签,整个过程会被重复两次生成一个最终的单倍型路径集合。
一个Rukki 特有的特征是,它可以在局部歧义和组装图的断点进行scaffold,不需要额外的scaffolding技术 。例如,当遇见bubble时,他会完整遍历或引入gap。另一个时在loop tangle的scaffolding,一般是由于串联重复引起的。这种情况下,gap的长度根据tangle内所有node的长度和以及overlap估计。如果一个单倍型是完整的,Rukki在scaffold另一个中断的单倍型,用完整的单倍型估计断开的单倍型上gap的长度。
Rukki是一个独立模块,只用组装图和node上特定单倍型的marker的数量作为输入。可以适配其他方法,但可能需要额外的调整。
Consensus
Verkko 主要的数据结构是同聚物压缩有的组装图,需要恢复node或者路径的基因组序列得到consensus。Verkko的node map用来翻译节点或者路径到MBG node的序列。MBG节点的序列和LA reads用来构建 最终图的每个节点的 layout,每个LA reads 分配给一个精确匹配最长的位置,如果多个一样,则都分配,ULreads只会在最初的LA graph上,需要填充gap时包含在layout中,然后使用Canu的模块生成layout的consensus,默认只有一个read支持的consensus序列会被裁剪掉。
Results
Verkko overview
受CHM13启发,它的方法是半人工,且不能直接用于杂合子 heterozygout。Verko拓展了这个策略到双倍体,并能完全自动执行这个过程。Verkko构建了一个高质量组装图,解决 repeat,在单倍型和重复之间探查微小变异。通过整合 UL reads 和 单倍型marker 简化这个图,填充gap和单倍型定型。
同聚物的插入删除是主要的测序错误。流程主要是在同聚物压缩后进行的,在最终生成consensus时恢复。压缩后,用LA reads构建 multiplex DBG,比对 UL reads 填充gap 解决重复和单倍型,然后清理图,通过单倍型特异marker识别单倍型路径,恢复同聚物计算consensus。
最终的输出时定向的父本和母本的单倍型,和高精度的组装图。因为现有数据不能完全的将染色体组装成一个contig,Verkko利用组装图结构生成 TtoT (端粒到端粒),单倍型解决的 scaffold,所以移除了独立,错误 scaffold的步骤。
思考
整体和 bacteria 组装有点像,源节点和汇聚节点的方式可用,也可以打标签的方式。
,
